🧬 Blotting techniques
Blotting techniques.
This lesson explains the core ideas, methods, and exam-relevant applications for this topic in plant biotechnology. Focus on definitions, process steps, and practical uses for revision.
Blotting is the technique in which nucleic acids or proteins are immobilized onto a solid support
generally nylon or nitrocellulose membranes. Blotting of nucleic acid is the central technique for
hybridization studies. Nucleic acid labeling and hybridization on membranes have formed the
basis for a range of experimental techniques involving understanding of gene expression,
organization, etc.
Identifying and measuring specific proteins in complex biological mixtures, such as blood, have
long been important goals in scientific and diagnostic practice. More recently the identification of
abnormal genes in genomic DNA has become increasingly important in clinical research and
genetic counseling. Blotting techniques are used to identify unique proteins and nucleic acid
sequences. They have been developed to be highly specific and sensitive and have become
important tools in both molecular biology and clinical research.
General principle
The blotting methods are fairly simple and usually consist of four separate steps: electrophoretic
separation of protein or of nucleic acid fragments in the sample; transfer to and immobilization
on paper support; binding of analytical probe to target molecule on paper; and visualization of
bound probe. Molecules in a sample are first separated by electrophoresis and then transferred
on to an easily handled support medium or membrane. This immobilizes the protein or DNA
fragments, provides a faithful replica of the original separation, and facilitates subsequent
biochemical analysis. After being transferred to the support medium the immobilized protein or
nucleic acid fragment is localized by the use of probes, such as antibodies or DNA, that
specifically bind to the molecule of interest. Finally, the position of the probe that is bound to the
immobilized target molecule is visualized usually by autoradiography. Three main blotting
techniques have been developed and are commonly called Southern, northern and western
blotting.
Southern blot
Southern blot is a method used to check for the presence of a DNA sequence in a DNA sample.
The method is named after its inventor, the British biologist Edwin Southern.
The procedure for Southern blot technique is as detailed below:
Restriction endonucleases are used to cut high-molecular-weight DNA strands into smaller
fragments, which are then electrophoresed on an agarose gel to separate them by size.
If the DNA fragments are larger than 15 kb, then prior to blotting, the gel may be treated with
an acid, such as dilute HCl, which depurinates the DNA fragments, breaking the DNA into
smaller pieces, thus allowing more efficient transfer from the gel to membrane.
- If alkaline transfer methods are used, the DNA gel is placed into an alkaline solution
(containing NaOH) to denature the double-stranded DNA. The denaturation in an alkaline
environment may improve binding of the negatively charged DNA to a positively charged
membrane, separating it into single DNA strands for later hybridization to the probe and
destroys any residual RNA that may still be present in the DNA.
A sheet of nitrocellulose (or nylon) membrane is placed on top of (or below, depending on
the direction of the transfer) the gel. Pressure is applied evenly to the gel (either using
suction, or by placing a stack of paper towels and a weight on top of the membrane and
gel), to ensure good and even contact between gel and membrane. Buffer transfer by
capillary action from a region of high water potential to a region of low water potential
(usually filter paper and paper tissues) is used to move the DNA from the gel on to the
membrane; ion exchange interactions bind the DNA to the membrane due to the negative
charge of the DNA and positive charge of the membrane.
- The membrane is then baked in a vacuum or regular oven at 80 °C for 2 hours or exposed
to ultraviolet radiation (nylon membrane) to permanently attach the transferred DNA to the
membrane.
The membrane is then exposed to a hybridization probe—a single DNA fragment with a
specific sequence whose presence in the target DNA is to be determined. The probe DNA is
labelled so that it can be detected, usually by incorporating radioactivity or tagging the
molecule with a fluorescent or chromogenic dye.
- After hybridization, excess probe is washed from the membrane and the pattern of
hybridization is visualized on X-ray film by autoradiography in the case of a radioactive or
fluorescent probe, or by development of colour on the membrane if a chromogenic detection
method is used.
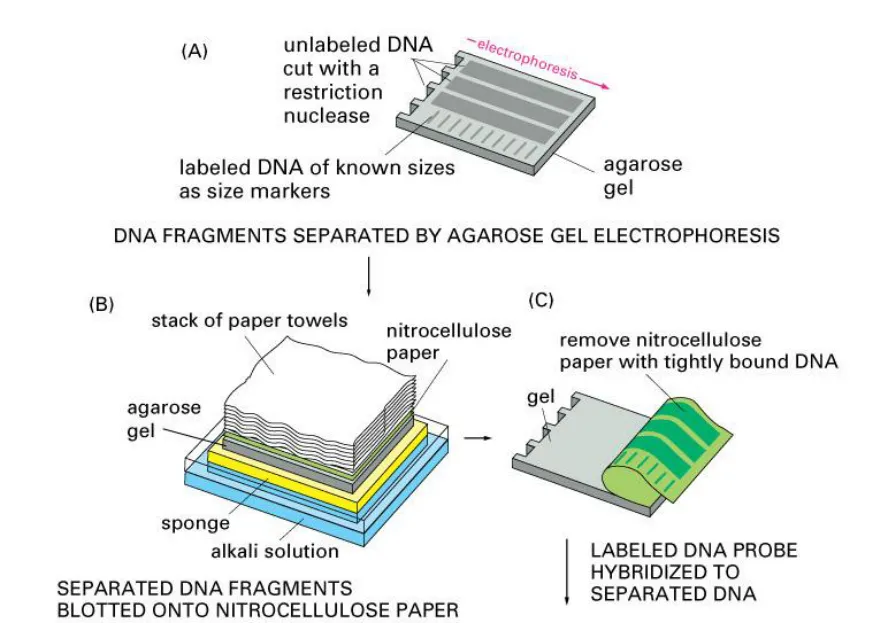
Hybridization of the probe to a specific DNA fragment on the filter membrane indicates that this
fragment contains DNA sequence that is complementary to the probe. The transfer step of the
DNA from the electrophoresis gel to a membrane permits easy binding of the labeled
hybridization probe to the size-fractionated DNA. Southern blots performed with restriction
enzyme-digested genomic DNA may be used to determine the number of sequences (e.g., gene
copies) in a genome. A probe that hybridizes only to a single DNA segment that has not been
cut by the restriction enzyme will produce a single band on a Southern blot, whereas multiple
bands will likely be observed when the probe hybridizes to several highly similar sequences
(e.g., those that may be the result of sequence duplication). Modification of the hybridization
conditions (ie, increasing the hybridization temperature or decreasing salt concentration) may
be used to increase specificity and decrease hybridization of the probe to sequences that are
less than 100% similar.
Northern blot
The northern blot technique is used to study gene expression by detection of RNA (or isolated
mRNA) in a sample. With northern blotting it is possible to observe cellular control over structure
and function by determining the particular gene expression levels during differentiation,
morphogenesis, as well as abnormal or diseased conditions. This technique was developed in
1977 by James Alwine, David Kemp and George Stark at Stanford University. Northern blotting
takes its name from its similarity to the first blotting technique, the Southern blot. The major
difference is that RNA, rather than DNA, is analyzed in the northern blot.
Procedure
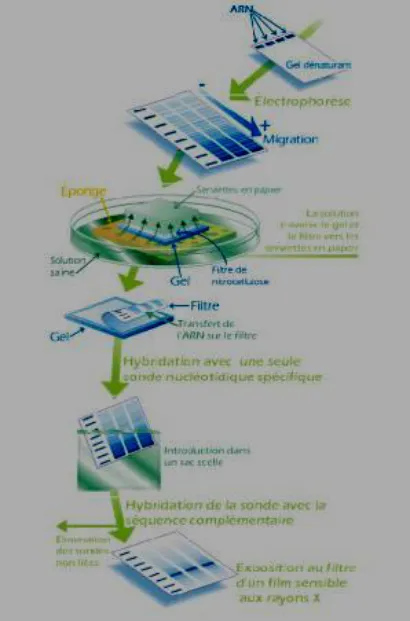
The blotting procedure starts with extraction of total RNA from a homogenized tissue sample.
The mRNA can then be isolated through the use of oligo (dT) cellulose chromatography to
maintain only those RNAs with a poly(A) tail. RNA samples are then separated by gel
electrophoresis. A nylon membrane with a positive charge is the most effective for use in
northern blotting since the negatively charged nucleic acids have a high affinity for them. The
transfer buffer used for the blotting usually contains formamide because it lowers the annealing
temperature of the probe-RNA interaction preventing RNA degradation by high temperatures.
Once the RNA has been transferred to the membrane it is immobilized through covalent linkage
to the membrane by UV light or heat. After a probe has been labeled, it is hybridized to the RNA
on the membrane. The membrane is washed to ensure that the probe has bound specifically.
The hybrid signals are then detected by X-ray film and can be quantified by densitometry.
Applications
Northern blotting allows in observing a particular gene's expression pattern between tissues,
organs, developmental stages, environmental stress levels, pathogen infection. The technique
has been used to show over expression of oncogenes and down regulation of tumor-suppressor
genes in cancerous cells when compared to 'normal' tissue, as well as the gene expression in
the rejection of transplanted organs. If an up regulated gene is observed by an abundance of
mRNA on the northern blot the sample can then be sequenced to determine if the gene is
known to researchers or if it is a novel finding. The expression patterns obtained under given
conditions can provide insight into the function of that gene. Since the RNA is first separated by
size, if only one probe type is used variance in the level of each band on the membrane can
provide insight into the size of the product, suggesting alternative splice products of the same
gene or repetitive sequence motifs. The variance in size of a gene product can also indicate
deletions or errors in transcript processing, by altering the probe target used along the known
sequence it is possible to determine which region of the RNA is missing.
Advantages & disadvantages
Analysis of gene expression can be done by several different methods including RT-PCR,
RNase protection assays, microarrays, serial analysis of gene expression (SAGE), as well as
northern blotting. Microarrays are quite commonly used and are usually consistent with data
obtained from northern blots, however at times northern blotting is able to detect small changes
in gene expression that microarrays cannot. The advantage that microarrays have over northern
blots is that thousands of genes can be visualized at a time while northern blotting is usually
looking at one or a small number of genes. A problem in northern blotting is often sample
degradation by RNases (both endogenous to the sample and through environmental
contamination) which can be avoided by proper sterilization of glassware and the use of RNase
inhibitors such as DEPC (diethylpyrocarbonate). The chemicals used in most northern blots can
be a risk to the researcher, since formaldehyde, radioactive material; ethidium bromide, DEPC,
and UV light are all harmful under certain exposures. Compared to RT-PCR northern blotting
has a low sensitivity but it also has a high specificity which is important to reduce false positive
results. The advantages of using northern blotting include the detection of RNA size, the
observation of alternate splice products, the use of probes with partial homology, the quality and
quantity of RNA can be measured on the gel prior to blotting, and the membranes can be stored
and reprobed for years after blotting.
Reverse northern blot
A variant of the procedure known as the reverse northern blot is occasionally used. In this
procedure, the substrate nucleic acid (that is affixed to the membrane) is a collection of isolated
DNA fragments, and the probe is RNA extracted from a tissue and radioactively labelled. The
use of DNA microarrays that have come into widespread use in the late 1990s and early 2000s
is more akin to the reverse procedure, in that they involve the use of isolated DNA fragments
affixed to a substrate, and hybridization with a probe made from cellular RNA. Thus the reverse
procedure, though originally uncommon, enabled northern analysis to evolve into gene
expression profiling, in which many (possibly all) of the genes in an organism may have their
expression monitored.
Western blot
The western blot (alternatively, immunoblot ) is used to detect specific proteins in a given
sample of tissue homogenate or extract. The method originated from the laboratory of George
Stark at Stanford. The name western blot was given to the technique by W. Neal Burnette.
Steps in a western blot
Tissue preparation
Samples may be taken from whole tissue or from cell culture. In most cases, solid tissues are
first broken down mechanically using a blender (for larger sample volumes), homogenizer
(smaller volumes) or sonication. Assorted detergents, salts and buffers may be employed to
encourage lysis of cells and to solubilize proteins. Protease and phosphatase inhibitors are
often added to prevent the digestion of the sample by its own enzymes. Tissue preparation is
often done at cold temperatures to avoid protein denaturing. A combination of biochemical and
mechanical techniques, including various types of filtration and centrifugation can be used to
separate different cell compartments and organelles.
Gel electrophoresis
The proteins of the sample are separated using gel electrophoresis. Separation of proteins may
be by isoelectric point (pI), molecular weight, electric charge or a combination of these factors.
SDS-PAGE (SDS polyacrylamide gel electrophoresis) maintains polypeptides in a denatured
state once they have been treated with strong reducing agents to remove secondary and tertiary
structure and thus allows separation of proteins by their molecular weight. Sampled proteins
become covered in the negatively charged SDS and move to the positively charged electrode
through the acrylamide mesh of the gel. Smaller proteins migrate faster through this mesh and
the proteins are thus separated according to size. The concentration of acrylamide determines
the resolution of the gel - the greater the acrylamide concentration the better the resolution of
lower molecular weight proteins. The lower the acrylamide concentration the better the
resolution of higher molecular weight proteins. Proteins travel only in one dimension along the
gel for most blots.
Samples are loaded into wells in the gel. One lane is usually reserved for a marker or ladder, a
commercially available mixture of proteins having defined molecular weights, typically stained
so as to form visible, coloured bands. When voltage is applied along the gel, proteins migrate
into it at different speeds. These different rates of advancement separate into bands within each
lane. It is also possible to use a two-dimensional (2-D) gel which spreads the proteins from a
single sample out in two dimensions. Proteins are separated according to isoelectric point (pH
at which they have neutral net charge) in the first dimension, and according to their molecular
weight in the second dimension.
Transfer
In order to make the proteins accessible to antibody detection, they are moved from within the
gel onto a nitrocellulose or polyvinylidene difluoride (PVDF) membrane similar to Southern blot
DNA transfer. Another method for transferring the proteins is called electro blotting and uses an
electric current to pull proteins from the gel into the PVDF or nitrocellulose membrane. The
proteins move from within the gel onto the membrane while maintaining the organization they
had within the gel. As a result of this "blotting" process, the proteins are exposed on a thin
surface layer for detection. Protein binding is based upon hydrophobic interactions, as well as
charged interactions between the membrane and protein. Nitrocellulose membranes are
cheaper than PVDF, but are far more fragile and do not stand up well to repeated probings.
The uniformity and overall effectiveness of transfer of protein from the gel to the membrane can
be checked by staining the membrane with Coomassie or Ponceau S dyes. Ponceau S is the
more common of the two, due to Ponceau S's higher sensitivity and its water solubility makes it
easier to subsequently destain and probe the membrane.
Blocking
Since the membrane has been chosen for its ability to bind protein and both antibodies and the
target are proteins, steps must be taken to prevent interactions between the membrane and the
antibody used for detection of the target protein. Blocking of non-specific binding is achieved by
placing the membrane in a dilute solution of protein - typically Bovine serum albumin (BSA) or
non-fat dry milk (both are inexpensive), with a minute percentage of detergent such as Tween
20. The protein in the dilute solution attaches to the membrane in all places where the target
proteins have not attached. Thus, when the antibody is added, there is no room on the
membrane for it to attach other than on the binding sites of the specific target protein. This
reduces "noise" in the final product of the Western blot, leading to clearer results, and eliminates
false positives.
Detection
During the detection process the membrane is "probed" for the protein of interest with a
modified antibody which is linked to a reporter enzyme, which when exposed to an appropriate
substrate drives a colourimetric reaction and produces a colour. For a variety of reasons, this
traditionally takes place in a two-step process, although there are now one-step detection
methods available for certain applications.
Two step
Antibodies are generated when a host species or immune cell culture is exposed to the protein
of interest. Normally, this is part of the immune response; whereas here they are harvested and
used as sensitive and specific detection tools that bind the protein directly.
After blocking, a dilute solution of primary antibody (generally between 0.5 and 5
micrograms/ml) is incubated with the membrane under gentle agitation. Typically, the solution is
composed of buffered saline solution with a small percentage of detergent, and sometimes with
powdered milk or BSA. The antibody solution and the membrane can be sealed and incubated
together for anywhere from 30 minutes to overnight. It can also be incubated at different
temperatures, with warmer temperatures being associated with more binding, both specific (to
the target protein, the "signal") and non-specific ("noise").
After rinsing the membrane to remove unbound primary antibody, the membrane is exposed to
another antibody, directed at a species-specific portion of the primary antibody. This is known
as a secondary antibody, and due to its targeting properties, tends to be referred to as "anti
mouse," "anti-goat," etc. Antibodies come from animal sources (or animal sourced hybridoma
cultures); an anti-mouse secondary will bind to just about any mouse-sourced primary antibody.
This allows some cost savings by allowing an entire lab to share a single source of mass
produced antibody, and provides far more consistent results. The secondary antibody is usually
linked to biotin or to a reporter enzyme such as alkaline phosphatase or horseradish
peroxidase. This means that several secondary antibodies will bind to one primary antibody and
enhance the signal.
Most commonly, a horseradish peroxidase-linked secondary is used in conjunction with a
chemiluminescent agent, and the reaction product produces luminescence in proportion to the
amount of protein. A sensitive sheet of photographic film is placed against the membrane, and
exposure to the light from the reaction creates an image of the antibodies bound to the blot. A
cheaper but less sensitive approach utilizes a 4-chloronaphthol stain with 1% hydrogen
peroxide; reaction of peroxide radicals with 4-chloronaphthol produces a dark brown stain that
can be photographed without using specialized photographic film.
As with the ELISPOT and ELISA procedures, the enzyme can be provided with a substrate
molecule that will be converted by the enzyme to a colored reaction product that will be visible
on the membrane (see the figure below with blue bands).
A third alternative is to use a radioactive label rather than an enzyme coupled to the secondary
antibody, such as labeling an antibody-binding protein like Staphylococcus Protein A with a
radioactive isotope of iodine. Since other methods are safer, quicker and cheaper this method is
now rarely used.
One step
Historically, the probing process was performed in two steps because of the relative ease of
producing primary and secondary antibodies in separate processes. This gives researchers and
corporations huge advantages in terms of flexibility, and adds an amplification step to the
detection process. Given the advent of high-throughput protein analysis and lower limits of
detection, however, there has been interest in developing one-step probing systems that would
allow the process to occur faster and with less consumables. This requires a probe antibody
which both recognizes the protein of interest and contains a detectable label, probes which are
often available for known protein tags. The primary probe is incubated with the membrane in a
manner similar to that for the primary antibody in a two-step process, and then is ready for direct
detection after a series of wash steps.
Analysis
After the unbound probes are washed away, the western blot is ready for detection of the
probes that are labeled and bound to the protein of interest. In practical terms, not all westerns
reveal protein only at one band in a membrane. Size approximations are taken by comparing
the stained bands to that of the marker or ladder loaded during electrophoresis. The process is
repeated for a structural protein, such as actin or tubulin, that should not change between
samples. The amount of target protein is indexed to the structural protein to control between
groups. This practice ensures correction for the amount of total protein on the membrane in
case of errors or incomplete transfers.
Colorimetric detection
The colorimetric detection method depends on incubation of the western blot with a substrate
that reacts with the reporter enzyme (such as peroxidase) that is bound to the secondary
antibody. This converts the soluble dye into an insoluble form of a different color that
precipitates next to the enzyme and thereby stains the membrane. Development of the blot is
then stopped by washing away the soluble dye. Protein levels are evaluated through
densitometry (how intense the stain is) or spectrophotometry.
Chemiluminescent detection
Chemiluminescent detection methods depend on incubation of the western blot with a substrate
that will luminesce when exposed to the reporter on the secondary antibody. The light is then
detected by photographic film, and more recently by CCD cameras which captures a digital
image of the western blot. The image is analysed by densitometry, which evaluates the relative
amount of protein staining and quantifies the results in terms of optical density. Newer software
allows further data analysis such as molecular weight analysis if appropriate standards are
used.
Radioactive detection
Radioactive labels do not require enzyme substrates, but rather allow the placement of medical
X-ray film directly against the western blot which develops as it is exposed to the label and
creates dark regions which correspond to the protein bands of interest (see image to the right).
The importance of radioactive detections methods is declining, because it is very expensive,
health and safety risks are high and ECL provides a useful alternative.
Fluorescent detection
The fluorescently labeled probe is excited by light and the emission of the excitation is then
detected by a photosensor such as CCD camera equipped with appropriate emission filters
which captures a digital image of the western blot and allows further data analysis such as
molecular weight analysis and a quantitative western blot analysis. Fluorescence is considered
to be among the most sensitive detection methods for blotting analysis.
Secondary probing
One major difference between nitrocellulose and PVDF membranes relates to the ability of each
to support "stripping" antibodies off and reusing the membrane for subsequent antibody probes.
While there are well-established protocols available for stripping nitrocellulose membranes, the
sturdier PVDF allows for easier stripping, and for more reuse before background noise limits
experiments. Another difference is that, unlike nitrocellulose, PVDF must be soaked in 95%
ethanol, isopropanol or methanol before use. PVDF membranes also tend to be thicker and
more resistant to damage during use.
2-D Gel Electrophoresis
2-dimensional SDS-PAGE uses the principles and techniques outlined above. 2-D SDS-PAGE,
as the name suggests, involves the migration of polypeptides in 2 dimensions. For example, in
the first dimension polypeptides are separated according to isoelectric point, while in the second
dimension polypeptides are separated according to their molecular weight. The isoelectric point
of a given protein is determined by the relative number of positively (e.g. lysine and arginine)
and negatively (e.g. glutamate and aspartate) charged amino acids, with negatively charged
amino acids contributing to a high isoelectric point and positively charged amino acids
contributing to a low isoelectric point. Samples could also be separated first under nonreducing
conditions using SDS-PAGE and under reducing conditions in the second dimension, which
breaks apart disulfide bonds that hold subunits together. SDS-PAGE might also be coupled with
urea-PAGE for a 2-dimensional gel.
In principle, this method allows for the separation of all cellular proteins on a single large gel. A
major advantage of this method is that it often distinguishes between different isoforms of a
particular protein - e.g. a protein that has been phosphorylated (by addition of a negatively
charged group). Proteins that have been separated can be cut out of the gel and then analysed
by mass spectrometry, which identifies the protein.
Eastern blotting
It is a technique to detect protein post translational modification and is an extension of the
biochemical technique of western blotting. Proteins blotted from two dimensional SDS-PAGE
gel on to a PVDF or nitrocellulose membrane are analyzed for post-translational protein
modifications using probes specifically designed to detect lipids, carbohydrate, phospho-
moieties or any other protein modification.
The technique was developed to detect protein modifications in two species of Ehrlichia - E.
muris and IOE. Cholera toxin B subunit (which detects lipids), Concanavalin A (which detects
glucose moieties) and nitrophospho molybdate-methyl green (detects phosphoproteins) were
used to detect protein modifications. The technique showed that the antigenic proteins of the
non-virulent E.muris are more post-translationally modified than the highly virulent IOE.
The technique was conceptualized by S. Thomas while working on sandal spike phytoplasma
and developed at the Dept. of Pathology, University of Texas Medical Branch, Galveston,
Texas, while working on the intracellular bacteria, Ehrlichia.
Significance
Most of the proteins that are translated from mRNA undergo modifications before becoming
functional in cells. The modifications collectively, are known as post-translational modifications
(PTMs). The nascent or folded proteins, which are stable under physiological conditions, are
then subjected to a battery of specific enzyme-catalyzed modifications on the side chains or
backbones.
Post-translational protein modifications includes: acetylation, acylation (myristoylation,
palmitoylation), alkylation, arginylation, biotinylation, formylation, glutamylation, glycosylation,
glycylation, hydroxylation, isoprenylation, lipoylation, methylation, nitroalkylation,
phosphopantetheinylation, phosphorylation, prenylation, selenation, S-nitrosylation, sulfation,
transglutamination and ubiquitination (sumoylation).
Post-translational modifications occurring at the N-terminus of the amino acid chain play an
important role in translocation across biological membranes. These include secretory proteins in
prokaryotes and eukaryotes and also proteins that are intended to be incorporated in various
cellular and organelle membranes such as lysosomes, chloroplast, mitochondria and plasma
membranes. Expression of post translated proteins is important in several diseases.
Applications of Blotting and Hybridization Techniques
- Southern blotting technique is widely used to find specific nucleic acid sequence present in
different plant species.
- Northern blotting technique is widely used to find gene expression and regulation of specific
genes.
-
By using blotting technique we can identify infectious agents present in the sample.
-
We can identify inherited disease.
-
It can be applied to mapping restriction sites in single copy gene.
Disadvantages of Blotting and Hybridization Techniques
-
The process is a complex, cumbersome and time consuming one.
-
It requires electrophoretic separation.
-
Only one gene or RNA can be analysed at a time.
-
Gives information about presence of DNA, RNA or proteins but does not give information
about regulation and gene interaction.
Dot Blotting Techniques - The drawbacks of blotting techniques have lead to the development
of dot blotting technique which is more advanced, less time consuming, accurate and applicable
to a wide variety of gene/source simultaneously.
The dot or slot blotting technique is the most widely used of all techniques for analysing. None
of the blot methods require electrophoresis prior to blotting and hybridization. Hybridization of
cloned DNA without electrophoretic separation is called as dot blotting.
Plaque or Colony Blotting Techniques - This method was first developed by Granstiens and
Hogness (1975). This method is used to identify which colony of bacteria contains the DNA of
interest among thousands. In this procedure, the bacterial colonies to be screened are
transferred onto nitrocellulose or nylon membrane by using replica plating.
Due to the negative charge of the cell surface, some cells bind to the nitrocellulose membrane.
Then the membrane is placed in a solution of 0.5 N NaOH to break the cell surface, convert
dsDNA to ssDNA and to bind DNA to the membrane. Later, the membrane is transferred to a
solution containing protease solution after neutralizing with neutralization solution.
The DNA is fixed tightly to membrane by either W cross linking or oven baking. This membrane
is used for hybridization with a probe and analysed by using autoradiography or biotin method
for positive hybridization. A colony whose DNA print (as replica plating provides a replica print
master plate colony on the membrane) gives a positive hybridization can be picked from the
master plate.
Plaque blotting is similar to colony blotting; the only difference is that instead of bacterial colony,
a plaque is transferred onto the membrane. Benton and Davis developed this method in 1977.
The greatest advantage of this method is that several identical DNA prints can be easily made
from a single master plate containing bacteria/plaques which are to be made.
Dot Plot Assay Techniques - This method is widely used to hybridize DNA from a single cell
type against a wide variety of probes, for example, for a viral infection which cannot be identified
by normal conventional methods or if we want to know what all genes are expressed in a single
cell type (e.g. brain cell).
Cell type or cells that are to be screened are placed on the membrane as 'dot' in the order of
rows and columns. Then the cells are denatured by using enzymes or detergents (SDS) and
DNA is fixed by using W - cross link or oven baking. This membrane is then used for
hybridization by using probes (which are specific to a gene).
Questions
- The technique in which nucleic acids or proteins are immobilized onto a solid support is
called as ..…..
a). Blotting b). Hybridisation c ). Immobilization d). None of the above
- Blotting techniques are used to identify.................
a). Unique proteins b). Nucleic acid sequences c). Both a and b d). None of the above
- Blotting techniques consist of................. separate steps.
a). 4 b). 3 c). 5 d). None of the above
- Blotting techniques consist of................. separate steps.
a). 4 b). 3 c). 5 d). None of the above
- Southern blot is a method used to check for the presence of................
a). DNA b). RNA c). Protein d). None of the above
- Southern blot was invented by ................
a). Edwin Southern b). James Alwine c). David Kemp d). None of the above
- Northern blot was invented by ................
a). George Stark b). James Alwine c). David Kemp d). All the above
- Northern blot is a method used to check for the presence of................
a). DNA b). RNA c). Protein d). None of the above
- Northern blot is used................
a). Overexpression of oncogenes b). Downregulation of tumor-suppressor genes c). Both a and b d). None of the above
- The advantages of Northern blot are ................
a). Detection of RNA size b). Quality and quantity of RNA can be measured c) . Use of probes with partial homology d). All the above
- Immunoblot is the other name of ................
a). Northern blot b). Southern blot c). Western blot d). None of the above
- Western blot is a method used to check for the presence of................
a). DNA b). RNA c). Protein d). None of the above
- Southern blot was invented by ................
a). Edwin Southern b). James Alwine c). David Kemp d). Neal Burnette
- Eastern blot is the extension of ................
a). Northern blot b). Southern blot c). Western blot d). None of the above
- Eastern blot is a method used to check for the presence of................
a). DNA b). RNA c). Protein d). Protein post translational modification
- Eastern blot was invented by ................
a). Edwin Southern b). James Alwine c). Thomas d). Neal Burnette
- The disadvantages of blotting techniques include ................
a). Complex, cumbersome and time consuming process
a). Complex, cumbersome and time b). No information about regulation and consuming process gene interaction
c).Analysis of one gene or RNA at a d). All the above time
d). All the above
Summary Cheat Sheet
Quick Recall Points
- Define key terms in one line and revise their use in plant biotechnology.
- Memorize major steps, methods, and applications covered in this lesson.
- Practice exam-style distinctions between related concepts and techniques.
Exam Traps
- Do not confuse similar terms without checking context and biological level.
- Revise process order carefully; sequence-based questions are common.
- Link each method with its most likely application question.
References
1 source • [1]
References
Standard BSc Agriculture Plant Biotechnology notes
BookLesson Doubts
Ask questions, get expert answers